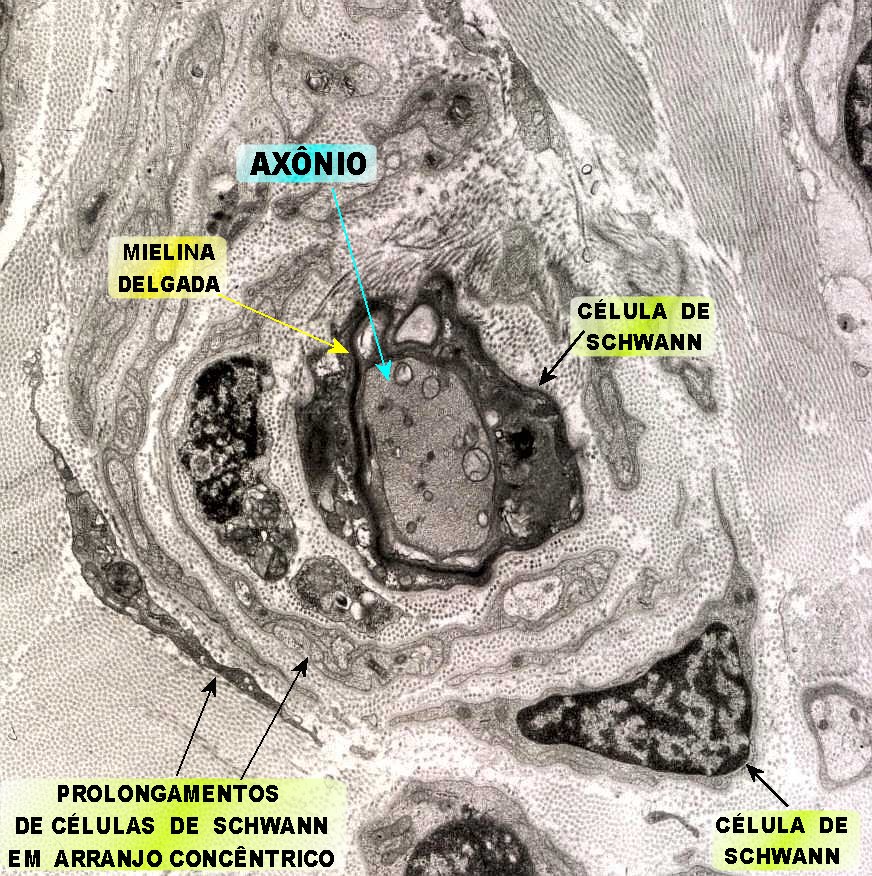
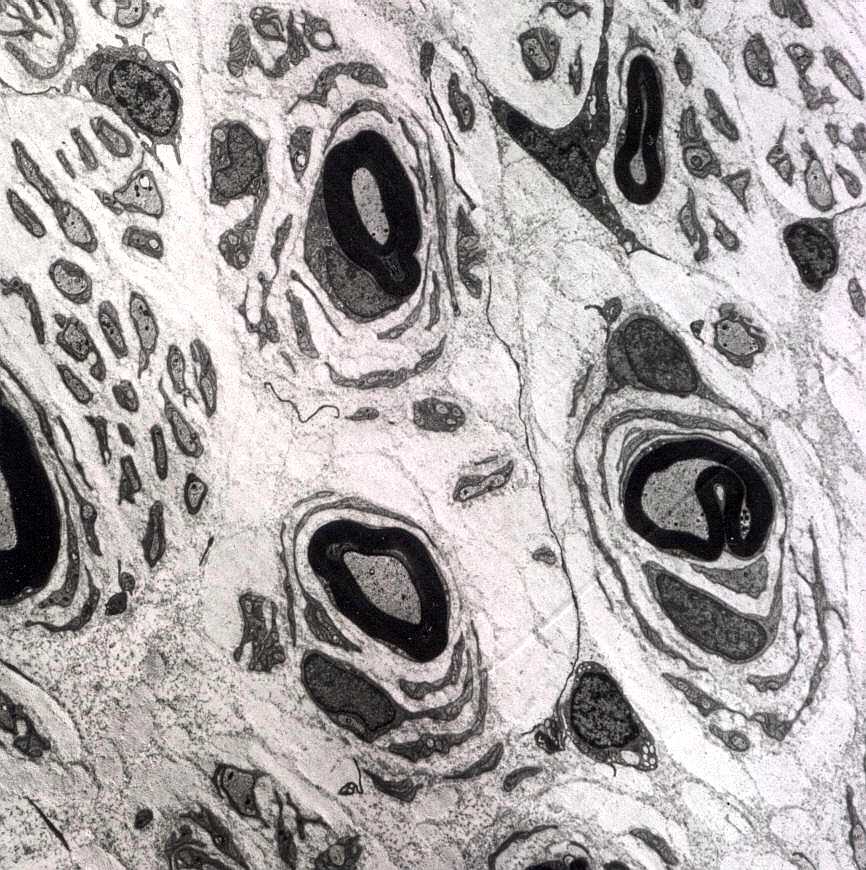
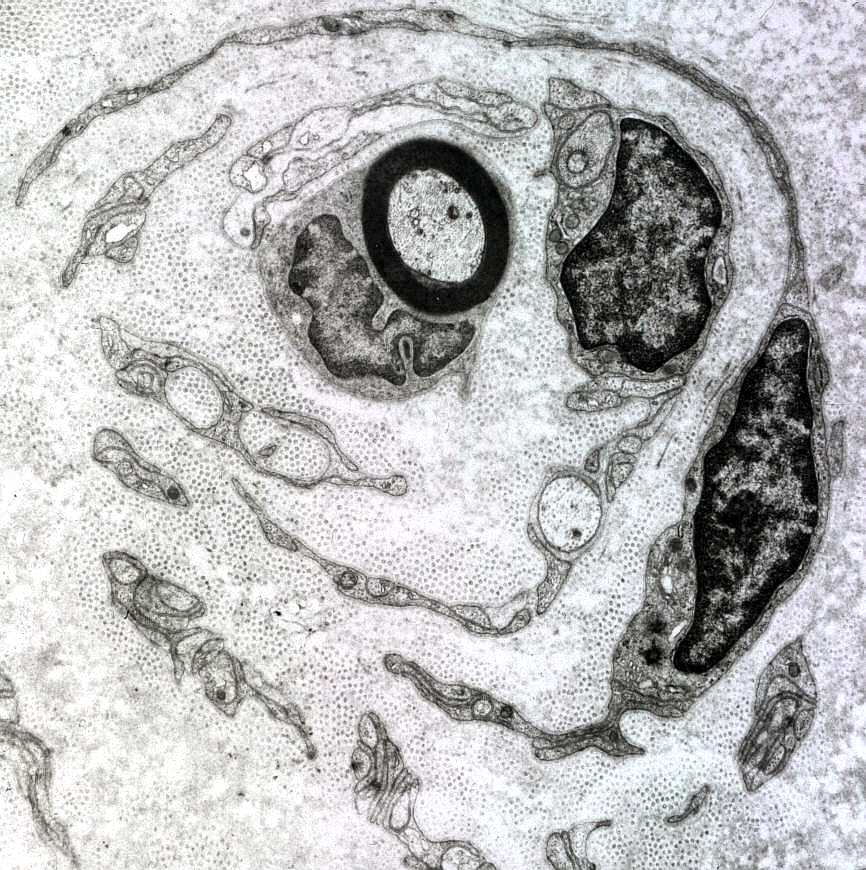
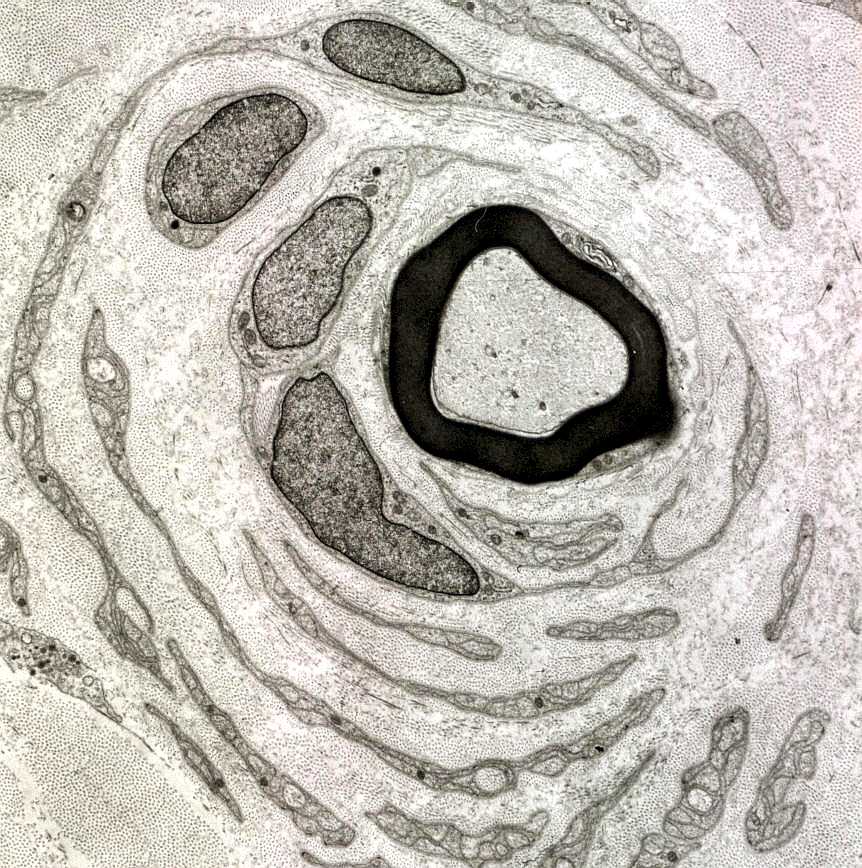
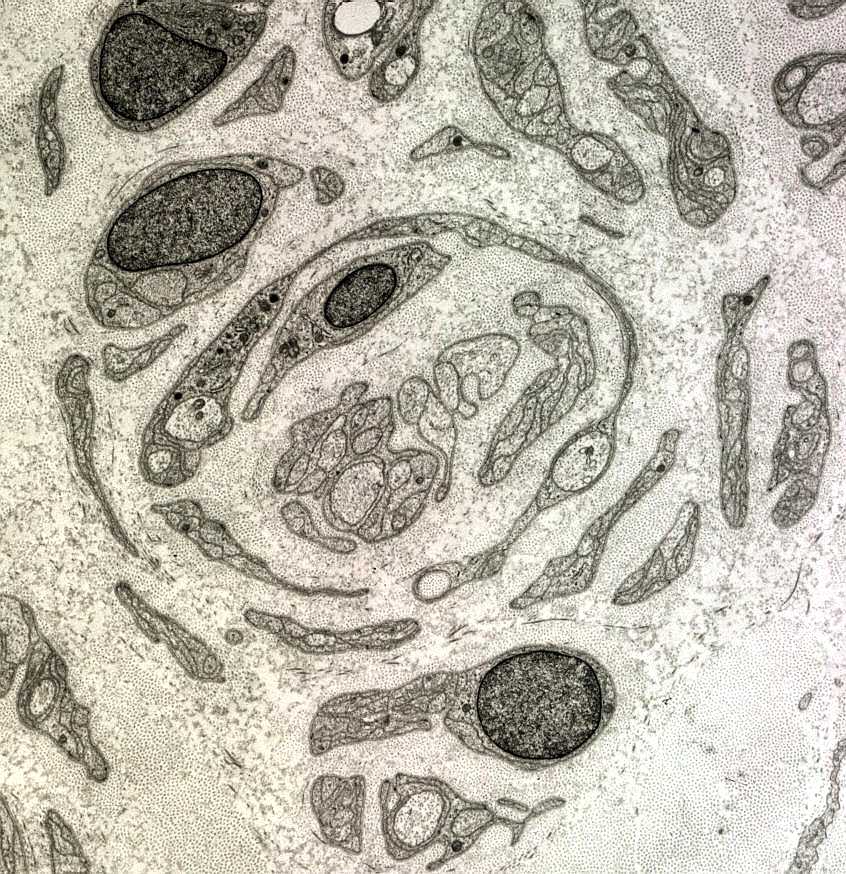
| A chamada neuropatia
hipertrófica resulta de repetidos ciclos de desmielinização
e remielinização em axônios periféricos.
Várias causas podem ser responsáveis. Entre elas, doenças
da mielina de fundo genético, em que proteínas da mielina
são defeituosas, como na doença de Déjérine-Sottas.
A mielina formada é frágil e degenera facilmente, sendo substituída
por uma nova, também frágil e assim por diante. No
processo, há proliferação dos prolongamentos das células
de Schwann, que tendem a dispor-se em volta dos axônios, à
maneira de pétalas de rosa ou em bulbo de cebola. As formações
concêntricas resultantes podem ser visíveis num corte transversal
de nervo incluído em parafina, já na HE.
Clinicamente,
há espessamento do nervo, que pode ser palpável, e até
visível, facilmente, como o radial, o ulnar e o grande auricular.
Citaremos como exemplos de neuropatia hipertrófica duas formas de
neuropatia hereditária sensitivo-motora (NHSM).
Doença de Charcot-Marie-Tooth
(ou atrofia muscular peroneal, ou NHSM tipo I).
Nesta forma,
a herança é autossômica dominante e o início
dos sintomas geralmente na 2a ou 3a décadas. O distúrbio
é predominantemente motor, caracterizando-se por atrofia progressiva
dos músculos distais dos membros inferiores. Há atrofia dos
pés (pés cavos) e das pernas, que se tornam desproporcionais
ao volume das coxas (relativamente poupadas), dando aspecto comparado a
pernas de cegonha ou a 'garrafa de champanhe invertida'. Mais tarde há
atrofia das mãos e antebraços. Ocorre também perda
de sensibilidade distal, porém discreta em relação
ao déficit motor. Atinge mais as modalidades proprioceptivas levando
a arreflexia profunda global. A velocidade de condução nervosa
medida por eletroneurografia está reduzida a 10-30 m/s (normal:
50-60 m/s em membro superior). A doença dura várias décadas
e pode parar de progredir espontaneamente.
Doença de Déjérine-Sottas
(ou NHSM tipo III). A herança é autossômica
recessiva. O início do quadro se dá na infância,
sendo a evolução mais rápida que no tipo I. Há
retardo no desenvolvimento neuromotor, fraqueza e atrofia dos membros na
porção distal, e ataxia. Os reflexos osteotendíneos
estão abolidos. Há anestesia e dores com distribuição
em bota e luva e perda do reflexo pupilar à luz. Muitos pacientes
desenvolvem deformidades como pé cavo e cifoscoliose, necessitando
cadeira de rodas. Ao exame físico, nota-se espessamento dos nervos,
que, na biópsia, mostram o quadro clássico de neuropatia
hipertrófica. As velocidades de condução nervosa estão
muito reduzidas.
|