|
|
Doença do Neurônio Motor - Texto |
|
|
|
Doença do Neurônio Motor - Texto |
|
| Esclerose
lateral amiotrófica (ELA)
Sinônimo doença do neurônio motor. Definição. Na doença do neurônio motor há degeneração primária (de causa desconhecida) dos neurônios motores superiores e inferiores, manifestando-se como fraqueza e atrofia dos músculos, associadas a sinais piramidais, como espasticidade e reflexos exaltados. Tipicamente, há preservação dos movimentos oculares e continência dos esfíncteres. A síndrome foi delineada por Charcot e Joffroy em 1869 e chamada por eles de esclerose lateral amiotrófica. O diagnóstico é baseado,além da clínica, em evidência eletrofisiológica de lesão dos neurônios do corno anterior e em casos mais difíceis, em biópsia muscular. Principais variantes
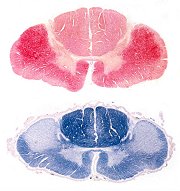
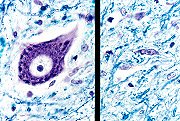
Epidemiologia. A distribuição da doença é mundial, com incidência mais ou menos uniforme de 2 casos novos por 100.000 habitantes/ano e prevalência de 5 : 100.000. Não há influência racial. Um possível aumento da incidência em tempos recentes poderia ser atribuído a maior longevidade das populações. Há preferência pelo sexo masculino 3:2. A idade média de início é 60 anos, e a duração entre 3-4 anos na forma com início apendicular e 2,5 anos na forma de início bulbar. Causas, possíveis mecanismos. A causa continua desconhecida, exceto em poucos casos familiais. Mais de 90 % dos casos são esporádicos. Entre as hipóteses em estudo estão excitotoxicidade, autoimunidade, perda de fatores de crescimento e infecções virais. Genética. Entre 5-10% dos pacientes têm uma forma familial autossômica dominante, com idade de início cerca de 10 anos antes da forma esporádica e tempo de evolução de 2 anos. Há ligação com o cromossomo 21q na região do gene da superóxido dismutase cobre/zinco (SOD1, SOD Cu/Zn). Foram encontradas cerca de 30 mutações pontuais do tipo missense. Contudo, tais mutações explicariam só 10 a 20% dos casos familiais. Como a SOD1 está implicada na remoção de radicais livres, inicialmente foi proposto que a mutação causasse dano aos neurônios via stress oxidativo (i.e. perda de função). Contudo, a atividade da enzima mutante pouco difere da normal. Por isso, aventou-se que a molécula mutante ganhe propriedades tóxicas (i.e. ganho de função). Mesmo dentro de uma família, a penetrância da mutação pode ser muito variável, com desde doença leve e curso prolongado a formas rapidamente progressivas. Neuropatologia. Para páginas ilustrativas, clique (1) (2). Macroscopicamente, em geral não se observam alterações no cérebro. Em alguns casos pode haver atrofia visível do giro pré-central. A medula espinal está atrófica e as raízes anteriores estão finas e acinzentadas, em comparação com as posteriores, que são normais. Histologicamente,
há perda dos neurônios motores do corno anterior, associada
a gliose reacional. Descrevem-se corpúsculos de inclusão
citoplasmáticos nos neurônios remanescentes. Os chamados
corpos de Bunina são inclusões pequenas e eosinófilas,
de 2 5 mm
cuja natureza é incerta. Há também inclusões
maiores que parecem-se com corpos de Lewy. Muitas destas inclusões
dão reação IH positiva para ubiquitina.
Extraído de
|
| Para imagens, clique » |
 |
 |
| Módulo Neuro - Página Inicial | Outros módulos | e-mails : [email protected]___[email protected] |